




News
Don’t get lost – How femtech can navigate the EU medical device and AI rules

By Xisca Borrás and Ellie Handy of the life sciences regulatory department at Bristows law firm
Femtech, short for female technology, is an important and fast growing sector. The EU is a key market for femtech, with five of the top 10 countries for femtech investment located in the EU.
Femtech products are developed for many areas of women’s health, such as menstrual health, pregnancy planning and monitoring, menopause and mental wellbeing.
As femtech is intrinsically linked to health needs, a key question for femtech products is whether they are regulated as medical devices or merely consumer products.
Additionally, many femtech products are embracing the use of artificial intelligence (“AI”). Therefore, another key question is whether products using AI will be regulated as “high-risk” AI systems under the EU’s new AI legal framework.
This article looks at when femtech apps and software qualify as medical devices in the EU and how the medical device and AI legal frameworks interact.
What is a software medical device?
The definition of “medical device” in the EU’s Medical Device Regulation 2017/745 (the “EU MDR”) includes software, used alone or in combination, that is intended by its legal manufacturer for a medical purpose. These medical purposes are listed in the EU MDR and include (amongst others):
- diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease;
- diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability; and
- control or support of conception.
The legal manufacturer is the person that puts their name/branding on the device, and takes responsibility for it.
Whether software is considered a medical device will depend on whether the manufacturer states it has a medical purpose in the relevant documentation/materials.
The EU MDR defines intended purpose as “the use for which a device is intended according to the data supplied by the manufacturer on the label, in the instructions for use or in promotional or sales materials or statements and as specified by the manufacturer in the clinical evaluation”.
What is the test for qualifying as a medical device in the EU?
There is a selection of guidance documents that can assist you in determining whether a product should qualify as a medical device. We summarise some of the key guidance below:
- MDCG 2019-11 rev.1
Under the EU MDR, the Medical Device Coordination Group (“MDCG”) has published guidance on the qualification and classification of software as a medical device. It sets out five decision steps to help determine if a piece of software is a medical device in the EU. The steps are:
- Step 1: Is the product software?
- Step 2: Is it standalone software (i.e., it is not an accessory nor driving/influencing the use of a hardware device) and does it not fall within Annex XVI?
- Step 3: Is it performing an action on data beyond storage, archival, communication, simple search or lossless compression?
- Step 4: Does it act for the benefit of an individual patient?
- Step 5: Does it have a medical purpose (as set out in the medical device definition)?
If the answer to all five questions is yes, it will qualify as a medical device. In this case, manufacturers will have to ensure they comply with the pre-market requirements set out in the EU MDR before they can place the software medical device on the market.
Notably, they will need to set up a qualify management system, compile a technical file, undergo the appropriate conformity assessment and affix a CE mark.
Importantly, the manufacturers would also need to consider post-market requirements, such as having a post-market surveillance system and undertaking post-market vigilance.
3. Other relevant guidance
The MDCG has also published a manual on borderline and classification of medical devices under the EU MDR.
Additional sources of guidance may also be available from national competent authorities. The legal manufacturer could also look at examples of other products already on the market to see how they are regulated (e.g. looking at EUDAMED). Although, we would caution anyone relying too heavily on the regulation of other products as there is no guarantee they are compliant.
What if you’re not a medical device?
If the software does not qualify as a medical device, the product will not have to comply with the EU MDR.
However, the manufacturer should be careful about how it promotes its product and the claims it makes about it because, as discussed above, a medical device is defined based on the manufacturer’s intended purpose.
Let’s take the example of a mere period app. Using it for logging period dates, tracking ovulation, and predicting future cycles has no medical purpose and is therefore not a medical device.
However, if its manufacturer recommends this piece of software for contraception and/or to support conception it will suddenly have a medical purpose and so, it would qualify as a medical device.
As such, the manufacturer would either have to bring the device into conformity with the EU MDR or take action to change the promotional materials to remove the medical claims.
Interaction between medical devices and AI legal frameworks
Under the EU MDR, devices are assigned risk classifications. For the lowest risk devices (Class I medical devices), the manufacturer can self-certify compliance with the EU MDR prior to the product being placed on the market or put into service in the EU.
However, high risk devices (Class IIa or above medical devices) must undergo a third party conformity assessment carried out by a notified body.
Notified body conformity assessments require a detailed review of the manufacturer’s quality management system, technical documentation, systems and procedures.
The process will often take more than a year to complete. Additionally, manufacturers have to grapple with ongoing burdens such as vigilance and post-market surveillance.
Under the EU MDR, most software as a medical device will be classified as a Class IIa or above.
Like the EU MDR, the EU’s Regulation (EU) 2024/1689 (the “AI Act”) also distinguishes between AI systems that pose different levels of risk.
The AI Act imposes onerous obligations on “high risk” AI systems, including in relation to accuracy, transparency, risk management, data quality and governance, and human oversight.
Although there is some overlap between the EU MDR and AI Act requirements, many are new AI-specific obligations. These pose a significant additional regulatory burden, increasing the complexity and cost of compliance for stakeholders.
Notably, the risk classification of an AI system that is itself, or is included in, a medical device is linked to the device’s classification under the EU MDR. Under the AI Act, AI systems are classified as “high risk” systems if:
(a) the AI system is a safety component of a medical device or the AI system itself is a medical device; and
(b) the medical device is required to undergo a third-party conformity assessment under the EU MDR.
Therefore, low risk medical devices (i.e., Class I medical devices) that are self-certified cannot be “high risk” AI systems.
Whereas, any device that requires a notified body to perform its conformity assessment will be a “high risk” AI system, and so will be subject to the additional AI Act requirements.
Unfortunately for those wishing to avoid the “high risk” AI system requirements, there are relatively few Class I devices under the EU MDR.
Therefore, the majority of medical devices that are an AI system or have an AI system as a safety component will qualify as a “high risk” AI system.
One notable example of a Class I device is software intended to support conception by calculating the user’s fertility status based on a validated statistical algorithm.
If this kind of software medical device is also an AI system, it would not be classed as a “high risk” AI system, so it would not be subject to the more onerous requirements in the AI Act.
However, the manufacturers of these devices would need to carefully consider any product developments that add additional functionality, as this can impact the risk classification of the product under both the EU MDR and AI Act.
For example, if the manufacturer added functionality to the Class I device so it could also be used as a means of contraception, it would become a Class IIb medical device and would need a third party conformity assessment.
In turn, as the software is also an AI system, this would mean the AI system would be considered “high-risk” and be subject to additional regulatory requirements under the AI Act.
Whilst AI has the potential to provide tremendous benefits for femtech, it also triggers additional complexity that can be time-consuming and costly to navigate.
It is important to get it right in terms of compliance in order to maintain consumer trust, avoid regulatory penalties, and pave the way for long-term success and viability.
By Xisca Borrás, Partner – Life sciences regulatory and Ellie Handy, Senior Associate – Life sciences regulatory at Bristows law firm.

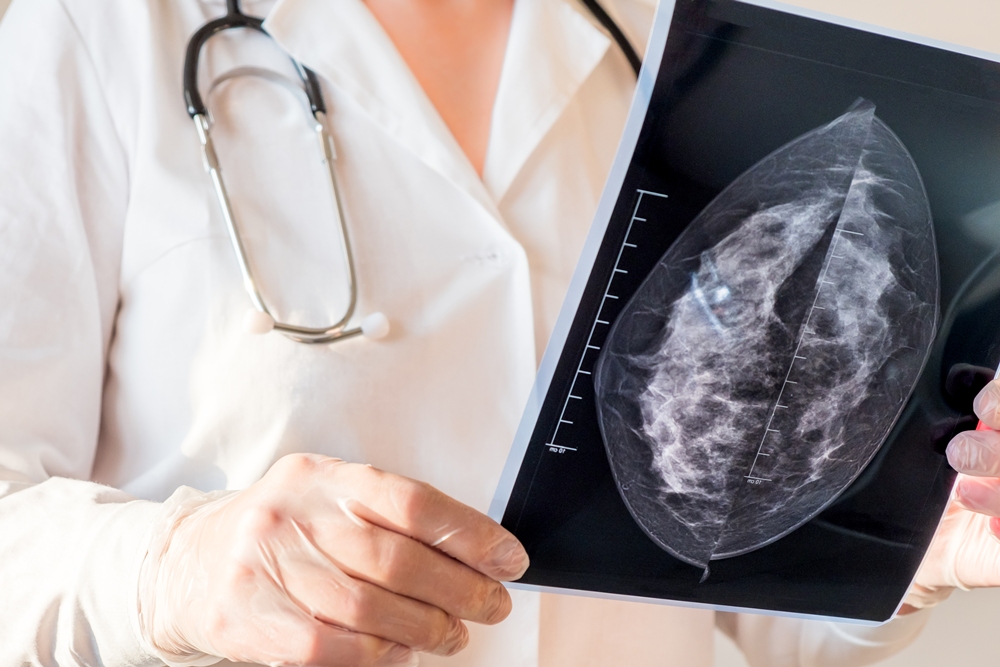
Changes in AI mammogram scores may help predict breast cancer years before diagnosis, research involving more than 54,000 women suggests.
Scores rose steadily among women who later developed the disease but remained broadly stable among those who did not.
The increase could be detected up to six years before diagnosis and became much steeper during the final two years.

Researchers led by Professor Constance Lehman, of Harvard Medical School and healthcare technology company Clairity, analysed screening mammograms taken between 2009 and 2019.
They used a validated, open-source deep learning model to calculate five-year breast cancer risk scores from the images alone.
Deep learning is a form of artificial intelligence trained to recognise complex patterns in large amounts of data.
The model examined the whole mammogram rather than relying on a limited, predetermined feature such as breast density.
Models of this kind have performed better than traditional risk models and breast density alone when estimating a woman’s five-year breast cancer risk.
The study initially included 239,703 consecutive two-dimensional screening mammograms from 89,882 patients across six imaging sites spanning urban tertiary, community-based and rural settings.
All were standard bilateral full-field digital mammography examinations, taken with or without digital breast tomosynthesis.
Digital breast tomosynthesis uses multiple low-dose X-ray images to create a three-dimensional view of the breast.
After exclusions, the final analysis involved 54,014 women with a median age of 61 and a total of 158,807 mammograms.
Each woman contributed one index examination and up to six previous annual mammograms. Women had a median of three scans each.
For women who developed cancer, the index examination was their final screening mammogram within the year before diagnosis. For the cancer-free group, it was their final mammogram during the five-year study period.
The model did not use demographic information, clinical records or historical imaging data when calculating each score.
Of the women included, 817, or one per cent, were diagnosed with breast cancer within 365 days of their index examination.
This included 451 women, or 55 per cent, with invasive breast cancer and 118, or 14 per cent, with ductal carcinoma in situ, known as DCIS.
DCIS occurs when abnormal cells are found inside a milk duct but have not spread into the surrounding breast tissue.
The cancer type was unknown for the remaining 248 patients, representing 30 per cent of the cancer group.
A total of 682 cancers, or 83 per cent, were detected through screening, while 135, or 17 per cent, were interval cancers diagnosed between routine mammograms.
The other 53,197 women were not diagnosed with breast cancer during follow-up and formed the cancer-free comparison group.
Professor Lehman said: “We observed clinically relevant differences in risk trajectories between women who did and did not develop cancer. The increase in scores among cancer patients was detectable as early as six years prior to diagnosis and became more pronounced over time.”
Among women later diagnosed with the disease, the median score rose from 2.1 five to six years before diagnosis to 6.6 at the index examination.
Scores among cancer-free women remained stable, with median values ranging from 1.8 to 2.2 throughout the study.
The rise among women who developed cancer was steepest during the two years before their index examination.
Professor Lehman said: “These findings demonstrate signals, invisible to the human eye, in the image alone can predict future risk. This is exciting, because 85 per cent of women diagnosed with breast cancer do not have a significant family history of breast cancer or known genetic mutations.”
Most breast cancers are considered sporadic, meaning they are not driven by inherited genetic changes or a family history of the disease.
Traditional risk models have a limited ability to distinguish between women who will and will not develop breast cancer when used across large screening populations.
Researchers said tracking how scores change over time could provide more information than calculating risk at a single appointment.
Professor Lehman said: “AI-derived risk scores can identify patients who are otherwise predisposed to the disease, and our findings demonstrate that image-based AI risk scores evolve over time and that changes in those scores may provide additional information about future breast cancer risk.”
The patterns remained consistent when women were grouped by age and breast density.
Breast density describes the amount of fibrous and glandular tissue visible on a mammogram. Dense tissue can make cancers harder to detect and is also associated with an increased risk of the disease.
Researchers said image-based scores could support personalised screening and risk-reduction strategies without relying on self-reported or inconsistent clinical information.
Professor Lehman said: “These trends remained robust across subgroups defined by age and breast density, further supporting the generalisability of our findings. This is particularly relevant given persistent disparities in screening performance across patient populations. A dynamic biomarker approach grounded in the imaging data could mitigate some of these disparities by enabling risk-based personalisation that does not rely on self-reported or inconsistent clinical data.”
A biomarker is a measurable sign that can indicate a person’s health, disease risk or response to treatment.
Changing scores could eventually help clinicians identify women who may benefit from additional imaging or measures intended to reduce their risk.
Professor Lehman said: “With the power of AI, computer vision, and the ability to extract predictive data, we are able to apply the power of imaging to risk assessment and preventing disease from developing. Having a dynamic risk score opens up a whole new domain of more effective preventive therapies for breast cancer, similar to how we screen for and treat patients with high cholesterol and hypertension.”
AI image-based risk scores are included in the 2026 National Comprehensive Cancer Network guidelines.
The guidelines recommend that, from the age of 35, women with an elevated five-year risk score of more than 1.7 per cent consider breast MRI alongside annual mammography.
An AI image-based model approved by the US Food and Drug Administration is already being used to calculate five-year breast cancer risk at selected US healthcare institutions.

Women’s health startup Ovum has raised US$4m in seed funding to develop its AI health journal and expand research using women’s health data.
The round valued the Melbourne startup at US$18m.
Ovum plans to use the funding to develop its artificial intelligence technology and longitudinal datasets, which track health information over time to reveal changes and patterns.

The AI captures symptoms, lifestyle factors, biometric measurements, reproductive health stages, medication, appointments and medical reports.
It uses this information to identify health patterns and create summaries and questions for medical appointments.
Ovum previously raised US$1.7m in pre-seed funding in February 2025 before launching its health journal app in August that year.
Since then, the company says the app has grown by 30 per cent month on month and recorded more than 20,000 downloads.
It has captured 57,000 health data insights and hosted more than 107,000 AI health conversations involving women aged between 15 and 84.
Founder Dr Ariella Heffernan-Marks developed the idea while she was a third-year medical student experiencing chronic migraines and was told that her pain was caused by anxiety.
The company describes the resulting women’s health journal as combining technology and clinical research to make health information more actionable and equitable for women.
Heffernan-Marks said: “I’ve sat on both sides of the desk, as a patient and as a doctor, and that’s why this mission matters so much to me.
“For too long, women have had to navigate healthcare systems that were not designed around their lived experiences or backed by sufficient female health data. Ovum exists to help women better understand their bodies, advocate for themselves with confidence, and contribute to research that improves care for future generations.”
Private health insurer Medibank is an Ovum partner, alongside Fernwood Fitness, Sweat and Menopause Friendly Australia.
Australian Red Cross Lifeblood is also involved in a pilot examining productivity losses caused by women reducing their working hours or leaving employment for health reasons.
Earlier in 2026, Ovum launched clinical trials with St George Hospital and the Royal Hospital for Women to assess AI as a preventative health tool for women.
The research is examining how women currently manage their health, which digital tools they use and whether AI could support health confidence, self-advocacy and continuity of care.
Continuity of care means receiving connected and consistent support across different appointments, healthcare professionals and services.
The funding round was led by Admiralty Capital Group, with participation from Antler, Giant Leap, Aviron Investments, Foggy Valley Aotearoa, Brisbane Angels and Think & Grow.
Existing investor LaunchVic, which is due to merge with Breakthrough Victoria, also participated through its Alice Anderson Fund, which focuses on female founders.
Amanda Andriano, founding partner at Admiralty Capital Group, said the gender health gap was a problem that should not be tolerated.
She said: “Ovum combines mission, market timing and technical capability with an exceptional founder uniquely positioned to lead this movement, and we believe that creates the foundation for a company of global significance.”

Women with endometriosis or painful periods were four to five times more likely to receive an STI diagnosis, a large Japanese study found.
Endometriosis occurs when tissue similar to the lining of the womb grows outside the womb. Although not strictly a menstrual disorder, it can cause pain, irregular periods and infertility.

The study was led by researchers at the University of Yamanashi and funded by Rohto Pharmaceutical Co.
The analysis examined health insurance claims from more than 3.4m women aged 40 or younger who had at least one healthcare visit during 2023.
Around 260,000 women, or 7.5 per cent of those included, had been diagnosed with endometriosis, dysmenorrhoea or both.
Dysmenorrhoea is the medical term for painful periods or menstrual cramps.
Women with endometriosis, dysmenorrhoea or both were four to five times more likely to have a recorded diagnosis of a sexually transmitted infection, or STI, than women without the conditions.
Diagnoses were significantly more common across every category examined, including chlamydia, gonorrhoea, trichomoniasis, genital herpes and other STIs.
Chlamydia was recorded in 3.5 per cent of women with menstruation-related conditions, compared with 0.7 per cent of those without them.
This represented a fivefold increase and the largest difference in prevalence between the two groups.
Gonorrhoea was diagnosed in 0.9 per cent of women with the conditions, compared with 0.2 per cent of those without them, also representing an increase of about five times.
Trichomoniasis, genital herpes and other STIs were diagnosed four to five times more often in women with endometriosis, dysmenorrhoea or both.
Women with endometriosis had the highest STI diagnosis rates overall.
Almost five per cent had a recorded chlamydia diagnosis, making it the most common STI in this group and more than seven times as prevalent as among women without menstruation-related conditions.
Women with dysmenorrhoea also had higher diagnosis rates for every STI included in the analysis.
The study found little evidence that hormonal treatments, including low-dose oestrogen-progestin therapy, affected STI diagnosis rates.
Differences between women who used hormonal treatment and those who did not were generally less than one percentage point.
Researchers suggested several possible explanations for the association between menstruation-related conditions and STI diagnoses.
One likely explanation is that women with endometriosis and dysmenorrhoea attend healthcare appointments more often.
As many STIs cause only mild symptoms, women seeking care more frequently for these conditions may be more likely to have infections detected.
Biological and behavioural factors may also play a part.
Menstruation-related conditions, particularly endometriosis, are associated with inflammation, pain during sex and sexual dysfunction, which could influence contraceptive practices and susceptibility to infection.
However, the authors said these possible explanations “remain speculative.”
They cautioned that differences in healthcare-seeking behaviour make it difficult to determine whether women with menstruation-related conditions acquire more infections or are simply more likely to receive a diagnosis.
The authors concluded that the findings underline the importance of STI screening and reproductive health education for women with endometriosis or painful periods.










 Menopause4 weeks ago
Menopause4 weeks agoPerimenopause misinformation ‘putting women at risk’

 Entrepreneur4 weeks ago
Entrepreneur4 weeks agoWomen’s Health Innovation Summit opens submissions for 2026 Innovation Showcase

 Insight3 weeks ago
Insight3 weeks agoBritish women among angriest in Europe, health survey reveals

 News4 weeks ago
News4 weeks agoWomen still being failed when they reach menopause, experts say

 Menopause2 weeks ago
Menopause2 weeks agoApple Health adds menopause and perimenopause tracking

 Menopause4 weeks ago
Menopause4 weeks agoSweden eyes domestic production of oestrogen patches amid menopause treatment shortage

 News2 weeks ago
News2 weeks agoFemtech World Awards 2026: Winners revealed

 News4 weeks ago
News4 weeks agoThree menopause innovators shortlisted for Femtech World Award
3 Comments