




Insight
Wearables could revolutionise pregnancy monitoring, study finds

Common fitness trackers may help track pregnancy complications by detecting heart rate changes that mirror hormonal shifts, new research suggests.
The study found devices such as Apple Watch, Garmin and Fitbit recorded heart rate patterns that aligned with key pregnancy hormones including oestrogen, progesterone and human chorionic gonadotropin (hCG).
These hormones play vital roles in supporting a healthy pregnancy.
Researchers analysed data from more than 5,600 people using PowerMom, a bilingual digital research platform.
From this group, 108 provided continuous data covering three months before conception through six months after giving birth.
Heart rate data showed distinct trends. Early pregnancy was marked by a small dip around weeks five to nine, followed by steady rises until about two months before delivery, peaking at up to 9.4 beats per minute above baseline.
After birth, heart rates dropped below pre-pregnancy levels before stabilising at around six months.
The researchers also tracked sleep and activity patterns throughout pregnancy.
Giorgio Quer is co-senior author and director of artificial intelligence at Scripps Research.
Quer said: “Wearable devices offer a unique opportunity to develop innovative solutions that address the high number of adverse pregnancy outcomes in the US.
“Our results show that signals collected via wearable sensors follow the expected changes in hormone levels and can detect unique patterns specific to live birth pregnancies, potentially allowing the monitoring of maternal health throughout the pregnancy and postpartum.”
To check the link between sensors and hormones, the team compared wearable heart rate data with published hormonal changes from previous studies, building models that predicted how heart rate should shift as hormone levels rose or fell.
In an exploratory analysis of a small number of cases, pregnancies ending in miscarriage or stillbirth showed different heart rate patterns compared with healthy pregnancies, though researchers said larger groups are needed to confirm this.
“Hormones play a key role in pregnancy outcomes,” explained co-senior author Tolúwalàṣẹ Àjàyí, principal investigator of PowerMom.
“Discovering the association between heart rate and hormone changes could unlock new ways to predict the beginning of pregnancy or identify signs of adverse outcomes such as gestational diabetes or preeclampsia.”
Preeclampsia is a condition involving high blood pressure during pregnancy, while gestational diabetes means raised blood sugar that develops in pregnancy.
Researchers say the approach could help provide continuous oversight for high-risk pregnancies, particularly in underserved communities.
Previous work has shown that wearable devices can identify infections such as COVID-19 and other health issues by detecting changes in body patterns.
The team plans further work to examine variations across different groups, regions and socioeconomic backgrounds.
They aim to build models that flag those who might need closer monitoring.
Future studies will test whether wearable data could directly guide clinical decisions.
Researchers also plan to gather both wearable and blood test data from the same participants to validate the link between hormones and heart rate.












Women’s Health Week Europe 2026 has released its full programme ahead of the October event at The Emirates Stadium in London on 7–8 October, with 700+ senior decision-makers and 80+ speakers confirmed across what will be the organisation’s most ambitious edition to date.
For the first time, the event will run across two dedicated stages, each built around a distinct set of questions facing the women’s health industry.
The Global Stage takes on the macro forces shaping the sector: where capital is flowing, how AI is transforming diagnosis and treatment, the gender data gap, wearable technology, stigmatised markets, and the policy landscape across Europe.
Confirmed speakers include Merete Clausen (EIF), Frida Polli (MIT), Nichole Young-Lin (Google), Alison Cave (MHRA), Emily Darlington MP, Kerry Buckley (Boots), Tim Davis (LSEG), Henriette Hessen (Verdane), Hillary Ball (Atomico), and Christine Hockley (British Business Bank).
The Scale Stage runs in parallel, focused on execution: how to navigate regulatory approval pathways, survive the valley of death, build the evidence stack that wins payers and partners, implement AI into a women’s health business, and position for acquisition. Sessions include a reverse pitch format, in which corporates and investors pitch to founders, and a founder’s guide to getting acquired.
The programme also includes two Pitch competitions, one per day, across the Consumer & Tech and Medical Devices & Therapeutics categories, with 16 finalists competing on the mainstage in front of the full delegate audience.
Every session is case study-driven, with speakers selected on the basis of having lived the problem they are on stage to solve.
Women’s Health Week Europe 2026 takes place 7–8 October at The Emirates Stadium, London. The full programme is available now.
View the 2026 programme here
Pre-agenda pricing ends 26 June
Tickets are currently available at pre-agenda pricing, with savings of up to £600 off standard pricing. The deadline is midnight on Friday 26 June. After that, prices go up.
Secure your place: https://wplatform.co/summits/womens-health-week-europe-2026?utm_source=advocacy&utm_medium=ext_email&utm_campaign=whw-europe-26-femtech-world#tickets
Also at The Emirates: Women’s Sport Summit 2026
The day before WHW Europe, on 6 October, The Emirates Stadium will also host the inaugural Women’s Sport Summit, a dedicated one-day event bringing together 400+ attendees from across sport, business, and investment. Focused on the commercial side of women’s sport, the Summit covers the full sports cycle: money, product, and market. Where women’s sport means business.

Most IVF add-ons lack reliable evidence, with benefits either absent or inconclusive, the largest review of its kind has found.
More than 70 per cent of IVF patients in the UK, Australia and New Zealand reportedly pay for one or more additional treatments.
However, researchers found that most of the procedures, medicines and techniques had no effect on fertility or were backed by limited or low-quality evidence.

Unproven add-ons can also lead to false hope, greater financial strain and unnecessary medical procedures at an already difficult time for patients.
Dr Sarah Lensen, of the University of Melbourne, said: “In many countries, infertility care is largely provided by private clinics where IVF is highly commercialised, and some add-ons are extremely expensive.
“Our review finds a lack of evidence that most of the IVF add-ons we assessed provide any benefit to patients. Unproven add-ons can lead to false hope, greater financial strain and unnecessary medical procedures at what already can be a very difficult time for patients.”
Researchers said concerns have grown in recent years about potentially untrustworthy randomised controlled trials in reproductive medicine, including studies of IVF add-ons.
The team set out to review the effectiveness and safety of 10 commonly offered add-ons using trustworthy studies.
Researchers initially identified 157 potentially eligible randomised controlled trials but excluded 72 because of concerns about their reliability.
Randomised controlled trials compare treatments by assigning participants to different groups, helping researchers assess whether an intervention causes a particular outcome.
The team combined data from the remaining 85 trials in a meta-analysis, which brings together findings from several studies.
The review found no effect on fertility or inconclusive evidence for seven of the 10 add-ons examined.
These included acupuncture, which involves inserting thin needles into points on the body, and corticosteroids, medicines that reduce inflammation and suppress immune activity.
Endometrial receptivity testing was also not backed by reliable evidence. The procedure involves taking a sample from the lining of the womb to examine patterns of gene activity.
Another add-on was intralipid infusion, which delivers a fat-containing liquid into the bloodstream.
Researchers separately examined injections of platelet-rich plasma into the ovaries and infusions of platelet-rich plasma into the womb.
Platelet-rich plasma is made from a patient’s blood and contains a high concentration of platelets, which play a role in healing.
The seventh treatment was pre-implantation genetic testing for aneuploidy, which examines embryos to check whether they have the expected number of chromosomes.
The review found only weak evidence of a possible benefit from three other add-ons.
EmbryoGlue, an embryo transfer medium containing hyaluronic acid, may increase the probability of pregnancy and live birth. However, the evidence on live birth rates was not considered robust.
Endometrial scratching, a minor procedure that deliberately disturbs the lining of the womb, may also increase the probability of pregnancy and live birth.
Physiological intracytoplasmic sperm injection, known as PICSI, selects sperm based on their ability to bind to hyaluronic acid. Weak evidence suggested it may reduce the risk of miscarriage.
Lensen said: “There is widespread misinformation about IVF add-ons with private clinic websites and patient forums on social media – major information sources for patients – often overstating the benefits and omitting the costs and risks of add-ons.
“IVF clinics and clinicians should carefully consider whether it is appropriate to offer unproven add-ons, as their availability is often perceived by patients as implicit endorsement of benefit.”
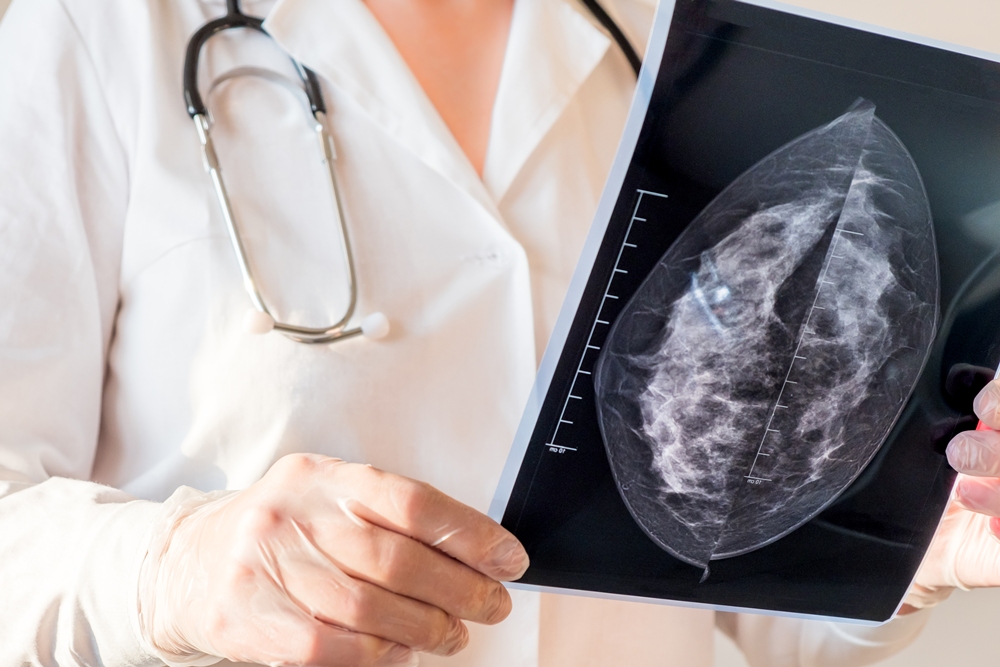
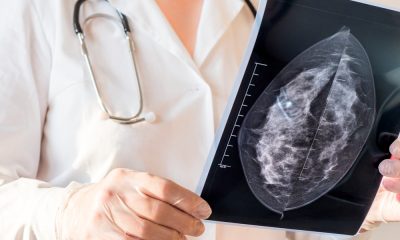
Changes in AI mammogram scores may help predict breast cancer years before diagnosis, research involving more than 54,000 women suggests.
Scores rose steadily among women who later developed the disease but remained broadly stable among those who did not.
The increase could be detected up to six years before diagnosis and became much steeper during the final two years.

Researchers led by Professor Constance Lehman, of Harvard Medical School and healthcare technology company Clairity, analysed screening mammograms taken between 2009 and 2019.
They used a validated, open-source deep learning model to calculate five-year breast cancer risk scores from the images alone.
Deep learning is a form of artificial intelligence trained to recognise complex patterns in large amounts of data.
The model examined the whole mammogram rather than relying on a limited, predetermined feature such as breast density.
Models of this kind have performed better than traditional risk models and breast density alone when estimating a woman’s five-year breast cancer risk.
The study initially included 239,703 consecutive two-dimensional screening mammograms from 89,882 patients across six imaging sites spanning urban tertiary, community-based and rural settings.
All were standard bilateral full-field digital mammography examinations, taken with or without digital breast tomosynthesis.
Digital breast tomosynthesis uses multiple low-dose X-ray images to create a three-dimensional view of the breast.
After exclusions, the final analysis involved 54,014 women with a median age of 61 and a total of 158,807 mammograms.
Each woman contributed one index examination and up to six previous annual mammograms. Women had a median of three scans each.
For women who developed cancer, the index examination was their final screening mammogram within the year before diagnosis. For the cancer-free group, it was their final mammogram during the five-year study period.
The model did not use demographic information, clinical records or historical imaging data when calculating each score.
Of the women included, 817, or one per cent, were diagnosed with breast cancer within 365 days of their index examination.
This included 451 women, or 55 per cent, with invasive breast cancer and 118, or 14 per cent, with ductal carcinoma in situ, known as DCIS.
DCIS occurs when abnormal cells are found inside a milk duct but have not spread into the surrounding breast tissue.
The cancer type was unknown for the remaining 248 patients, representing 30 per cent of the cancer group.
A total of 682 cancers, or 83 per cent, were detected through screening, while 135, or 17 per cent, were interval cancers diagnosed between routine mammograms.
The other 53,197 women were not diagnosed with breast cancer during follow-up and formed the cancer-free comparison group.
Professor Lehman said: “We observed clinically relevant differences in risk trajectories between women who did and did not develop cancer. The increase in scores among cancer patients was detectable as early as six years prior to diagnosis and became more pronounced over time.”
Among women later diagnosed with the disease, the median score rose from 2.1 five to six years before diagnosis to 6.6 at the index examination.
Scores among cancer-free women remained stable, with median values ranging from 1.8 to 2.2 throughout the study.
The rise among women who developed cancer was steepest during the two years before their index examination.
Professor Lehman said: “These findings demonstrate signals, invisible to the human eye, in the image alone can predict future risk. This is exciting, because 85 per cent of women diagnosed with breast cancer do not have a significant family history of breast cancer or known genetic mutations.”
Most breast cancers are considered sporadic, meaning they are not driven by inherited genetic changes or a family history of the disease.
Traditional risk models have a limited ability to distinguish between women who will and will not develop breast cancer when used across large screening populations.
Researchers said tracking how scores change over time could provide more information than calculating risk at a single appointment.
Professor Lehman said: “AI-derived risk scores can identify patients who are otherwise predisposed to the disease, and our findings demonstrate that image-based AI risk scores evolve over time and that changes in those scores may provide additional information about future breast cancer risk.”
The patterns remained consistent when women were grouped by age and breast density.
Breast density describes the amount of fibrous and glandular tissue visible on a mammogram. Dense tissue can make cancers harder to detect and is also associated with an increased risk of the disease.
Researchers said image-based scores could support personalised screening and risk-reduction strategies without relying on self-reported or inconsistent clinical information.
Professor Lehman said: “These trends remained robust across subgroups defined by age and breast density, further supporting the generalisability of our findings. This is particularly relevant given persistent disparities in screening performance across patient populations. A dynamic biomarker approach grounded in the imaging data could mitigate some of these disparities by enabling risk-based personalisation that does not rely on self-reported or inconsistent clinical data.”
A biomarker is a measurable sign that can indicate a person’s health, disease risk or response to treatment.
Changing scores could eventually help clinicians identify women who may benefit from additional imaging or measures intended to reduce their risk.
Professor Lehman said: “With the power of AI, computer vision, and the ability to extract predictive data, we are able to apply the power of imaging to risk assessment and preventing disease from developing. Having a dynamic risk score opens up a whole new domain of more effective preventive therapies for breast cancer, similar to how we screen for and treat patients with high cholesterol and hypertension.”
AI image-based risk scores are included in the 2026 National Comprehensive Cancer Network guidelines.
The guidelines recommend that, from the age of 35, women with an elevated five-year risk score of more than 1.7 per cent consider breast MRI alongside annual mammography.
An AI image-based model approved by the US Food and Drug Administration is already being used to calculate five-year breast cancer risk at selected US healthcare institutions.










 Menopause4 weeks ago
Menopause4 weeks agoPerimenopause misinformation ‘putting women at risk’

 Entrepreneur4 weeks ago
Entrepreneur4 weeks agoWomen’s Health Innovation Summit opens submissions for 2026 Innovation Showcase

 Insight3 weeks ago
Insight3 weeks agoBritish women among angriest in Europe, health survey reveals

 Menopause4 weeks ago
Menopause4 weeks agoWomen still being failed when they reach menopause, experts say

 Menopause2 weeks ago
Menopause2 weeks agoApple Health adds menopause and perimenopause tracking

 Menopause4 weeks ago
Menopause4 weeks agoSweden eyes domestic production of oestrogen patches amid menopause treatment shortage

 News2 weeks ago
News2 weeks agoFemtech World Awards 2026: Winners revealed

 News4 weeks ago
News4 weeks agoThree menopause innovators shortlisted for Femtech World Award